ABSTRACT
Objective: To evaluate the efficacy and safety of zuranolone, an investigational neuroactive steroid and GABAA receptor positive allosteric modulator, in major depressive disorder (MDD).
Methods: The phase 3, double-blind, randomized, placebo-controlled MOUNTAIN study enrolled adult outpatients with DSM-5–diagnosed MDD, 17-item Hamilton Depression Rating Scale total score (HDRS-17) ≥ 22, and Montgomery-Asberg Depression Rating Scale total score ≥ 32. Patients were randomized to treatment with zuranolone 20 mg, zuranolone 30 mg, or placebo for 14 days, followed by an observation period (days 15–42) and an extended follow-up (days 43–182). The primary endpoint was change from baseline (CFB) in HDRS-17 at day 15.
Results: 581 patients were randomized to receive zuranolone (20 mg, n = 194; 30 mg, n = 194) or placebo (n = 193). Day 15 HDRS-17 least-squares mean (LSM) CFB was −12.5 (zuranolone 30 mg) vs −11.1 (placebo; P = .116). Improvement vs placebo was significant at days 3, 8, and 12 (all P < .05). LSM CFB (zuranolone 20 mg vs placebo) was not significant at any measured time point. Post hoc analyses of zuranolone 30 mg in patients with measurable plasma zuranolone concentration and/or severe disease (baseline HDRS-17 ≥ 24) showed significant improvement vs placebo at days 3, 8, 12, and 15 (all P < .05). Incidence of treatment-emergent adverse events was similar between zuranolone and placebo groups; the most common (≥ 5%) were fatigue, somnolence, headache, dizziness, diarrhea, sedation, and nausea.
Conclusions: MOUNTAIN did not meet its primary endpoint. Significant rapid improvements in depressive symptoms were observed with zuranolone 30 mg at days 3, 8, and 12. Zuranolone was generally well tolerated in patients with MDD.
Trial Registration: ClinicalTrials.gov identifier: NCT03672175
J Clin Psychiatry 2023;84(2):22m14445
To cite: Clayton AH, Lasser R, Nandy I, et al. Zuranolone in major depressive disorder: results from MOUNTAIN—a phase 3, multicenter, double‐blind, randomized, placebo‐controlled trial. J Clin Psychiatry. 2023;84(2):22m14445.
To share: https://doi.org/10.4088/JCP.22m14445
© 2023 Physicians Postgraduate Press, Inc.
aUniversity of Virginia School of Medicine, Charlottesville, Virginia
bSage Therapeutics, Inc, Cambridge, Massachusetts
*Corresponding author: Anita H. Clayton, MD, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia School of Medicine, PO Box 800623, Charlottesville, VA 22908-0623 ([email protected]).
In the United States, an estimated 19.4 million adults experienced ≥ 1 major depressive episode in 20191; of these, 60% had severe functional impairment.2 Major depressive disorder (MDD) is one of the largest contributors to disability in the United States.2–6 Multiple genetic, epigenetic, and environmental risk factors contribute to the complex pathophysiology of MDD.7,8 Dysregulation of interconnected brain networks controlling mood is thought to give rise to depressive symptoms.9 In addition, disruptions in other key biological mechanisms, including neurotransmission,9,10 inflammation,10–12 and the stress response,10,12,13 may drive dysregulation in MDD neuronal networks. The excitatory-inhibitory balance in the brain is predominantly maintained by a balance between glutamatergic and GABAergic signaling, respectively.14,15 Alterations in GABA levels16–22 and GABAA receptor expression13,20,23–25 may contribute to the development of depression by disrupting this excitatory-inhibitory balance.19,26–28 GABAergic dysregulation in depression has been linked to altered stress response,12,13,20,29 increased levels of inflammation,11,12 and changes in neurotransmission.9,11,13,19,29
The goals of MDD treatment include improving quality of life by alleviating functional impairment, achieving complete remission of symptoms, and preventing relapse and recurrence.6,30,31 Standard-of-care (SOC) antidepressants, such as selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants, are used to treat approximately 75% of patients with MDD at diagnosis32; however, treatment response in many patients remains suboptimal. In the STAR*D study, approximately 37% of patients with MDD achieved remission after first-line treatment with citalopram; subsequent remission rates decreased with each additional trial of SOC antidepressant.33 Moreover, SOC therapies often require weeks or months to produce effects and long-term, chronic administration to be effective,33,34 potentially resulting in negative outcomes, including decreased likelihood of remission35–37 and nonadherence.33,34,38–41 Furthermore, SOC antidepressants are often associated with adverse effects, including insomnia, weight gain, and sexual dysfunction, which can lead to dose reduction, dose interruption, or nonadherence.38
Zuranolone is an investigational neuroactive steroid in clinical development as an oral, once-daily, 14-day treatment for MDD as part of the LANDSCAPE program and for the treatment of postpartum depression as part of the NEST program. These two programs include multiple studies examining use of zuranolone in several thousand people with a variety of doses, clinical endpoints, and treatment paradigms.1,42–45 While its exact mechanism is not fully elucidated, zuranolone is hypothesized to function as a positive allosteric modulator of GABAA receptors.44–50 Unlike benzodiazepines, a pharmacologic class thought to act via modulation of the synaptic GABAA receptor exclusively, zuranolone was shown in preclinical studies to modulate both synaptic and extrasynaptic GABAA receptors, potentiating both phasic and tonic postsynaptic currents, respectively.45,46 Zuranolone also has demonstrated synergistic phasic GABAA receptor activity with diazepam at different synaptic GABAA receptors, indicating a binding site distinct from benzodiazepines.46 Furthermore, while benzodiazepines decrease GABAA receptor surface expression, zuranolone has been shown to enhance GABAA receptor activity in a manner consistent with a sustained increase in cell surface expression of both synaptic and extrasynaptic GABAA receptors.46,51–53
Results from the MOUNTAIN study, a phase 3, double-blind, randomized, placebo-controlled clinical trial that assessed the efficacy, safety, and tolerability of zuranolone in adult outpatients with MDD, are reported here.
METHODS
Study Design
This study (MOUNTAIN; NCT03672175) was a randomized, double-blind, parallel-group, placebo-controlled, phase 3 trial in patients with MDD conducted at 55 sites across the United States (November 2018–March 2020) (for full list of study sites, see Supplementary Appendix 1). The study design comprised a screening period of ≤ 28 days, a 14-day treatment period, a 4-week observation period, and an extended follow-up period through day 182 (6 months) after the last dose of zuranolone (Supplementary Figure 1). Antidepressant use during the trial was permitted, provided participants were on a stable dose for at least 60 days prior to day 1 and agreed to continue on the stable dose through day 42. Initiation of new antidepressants or any other medications thought to have an impact on efficacy or safety endpoints was not allowed between screening and completion of the day 42 assessments. Eligible patients were stratified by the baseline use of antidepressants and randomly assigned in a 1:1:1 ratio to receive either zuranolone 20 mg, zuranolone 30 mg, or matching placebo. Patients self-administered a single oral dose daily in the evening with food, preferably fat-containing meals to increase absorption (Sage Therapeutics, data on file, 2019) for 14 days.
The study was performed in accordance with the ethical principles from the Declaration of Helsinki and was consistent with International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use and Good Clinical Practice guidelines, with approval from each site and written informed consent from each patient.
Dose Selection
Zuranolone 30 mg dose was selected based on a phase 2 study in patients with MDD.54 The lower, once-daily 20 mg dose was included to assess for minimal effective dose. Dose adjustments were not permitted.
Inclusion/Exclusion Criteria
Eligible patients were aged 18–65 years with a diagnosis of MDD (Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Clinical Trials Version [SCID-5-CT]) with symptoms present for ≥ 4 weeks and a 17-item Hamilton Depression Rating Scale total score55 (HDRS-17) ≥ 22. In a protocol amendment early in the study, the HDRS-17 requirement was replaced with the requirement of Montgomery-Asberg Depression Rating Scale total score56 (MADRS) ≥ 30 to reduce the potential for overrepresentation of insomnia items. In both measures, one can get up to 6 points for insomnia, but because MADRS has a higher total score, 6 points on the MADRS scale is less weighted than on the HDRS-17 scale. After this amendment, a blinded data review found that many patients had day 1 HDRS-17 < 22 (some as low as 13); consequently, the protocol was amended again to include patients with MADRS ≥ 32 and HDRS-17 ≥ 22 at screening and day 1 (prior to dosing) to better accrue patients with the intended severity of depression (modified full analysis set [mFAS]). Of the total 570 patients dosed, 271 had been enrolled at the time of the latter amendment (March 25, 2019).
Reasons for exclusion included attempted suicide associated with the current MDD episode; treatment-resistant depression, defined as persistent depressive symptoms despite treatment with adequate doses of 2 different classes of antidepressants within the current MDD episode (excluding antipsychotics) for at least 4 weeks; history of seizures, bipolar disorder, schizophrenia, and/or schizoaffective disorder; active psychosis; pregnant or within 4 weeks postpartum; and substance use disorder diagnosed within 12 months prior to screening. The full list of eligibility criteria is available in Supplementary Appendix 2.
Outcomes
The primary endpoint was change from baseline (CFB) in HDRS-17 at day 15. Secondary endpoints included CFB in Clinical Global Impression-Severity57 (CGI-S) at day 15; CFB in HDRS-17 and CFB in CGI-S at other measured time points (days 3, 8, 12, 21, 28, 35, 42, 70, 126, and 182); HDRS-17 response rate (≥ 50% reduction from baseline in HDRS-17; all measured time points); HDRS-17 remission rate (HDRS-17 ≤ 7; all measured time points); CFB in MADRS (all measured time points); Clinical Global Impression-Improvement57 (CGI-I) response rate (“much improved” or “very much improved”; all measured time points); and CFB in Hamilton Anxiety Rating Scale total score (HARS) at day 15 and all other measured time points. Prespecified exploratory endpoints included patient-reported outcomes (eg, Changes in Sexual Functioning Questionnaire short-form total score [CSFQ-14]58). Total CSFQ-14 scores ≤ 47 for females and ≤ 41 for males indicated sexual dysfunction. Plasma samples for pharmacokinetic analysis were collected at days 8 (± 1) and 15 (± 1).
Safety and tolerability were evaluated throughout the study by adverse event reporting, the Columbia-Suicide Severity Rating Scale (C-SSRS),59 the 20-item Physician Withdrawal Checklist (PWC-20; used to evaluate tolerance and dependence),60 and standard clinical assessments.
Statistical Analysis
A mixed-effects model for repeated measures was used for the analysis, including CFB in HDRS-17 at each visit as the dependent variable. Secondary and post hoc analyses were not adjusted for multiplicity; all reported P values for these analyses are nominal. The effect size—as measured using Cohen d for change in HDRS-17 scores from baseline at days 3, 8, 12, and 15, along with their corresponding 95% confidence intervals—was estimated using sample means and pooled standard deviations.
Logistic regression models for repeated measures using the generalized estimating equation method were applied for the analysis of HDRS-17 response, HDRS-17 remission, and CGI-I response. The post hoc analysis included 3 groups: patients with a HDRS-17 ≥ 24 at baseline, patients with any postbaseline plasma zuranolone concentration above quantification limit, and patients with a HDRS-17 ≥ 24 at baseline and any postbaseline plasma zuranolone concentration above quantification limit. Descriptive summary statistics are provided for other endpoints and for safety data (safety set; patients who received ≥ 1 dose of study drug).
Additional information regarding the statistical analysis plan is included in Supplementary Appendix 3.
RESULTS
Patient Disposition, Demographics, and Baseline Clinical Characteristics
A total of 581 patients were randomized; 570 (98.1%) received ≥ 1 dose of study drug (zuranolone 20 mg [n = 188], zuranolone 30 mg [n = 192], placebo [n = 190]; Figure 1). Less than 10% of patients discontinued during the 14-day treatment period (4.8% in the zuranolone 20-mg group, 7.8% in the zuranolone 30-mg group, and 7.9% in the placebo group). Overall, 157 patients (27.5%) discontinued the study, with consent withdrawal being the most common reason (15.4% [88/570]), followed by lost to follow-up (6.8%) and adverse events (2.1%).
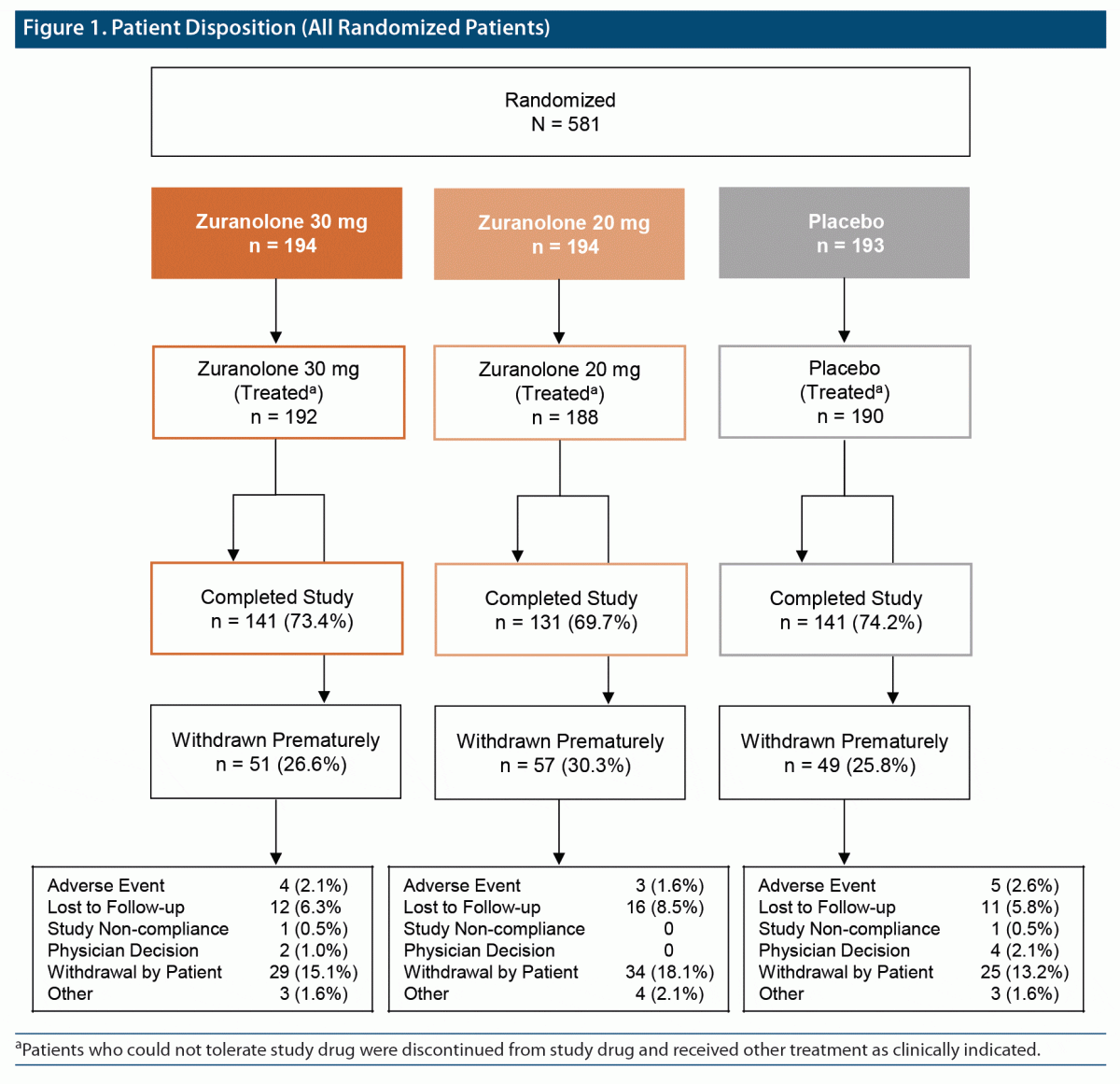
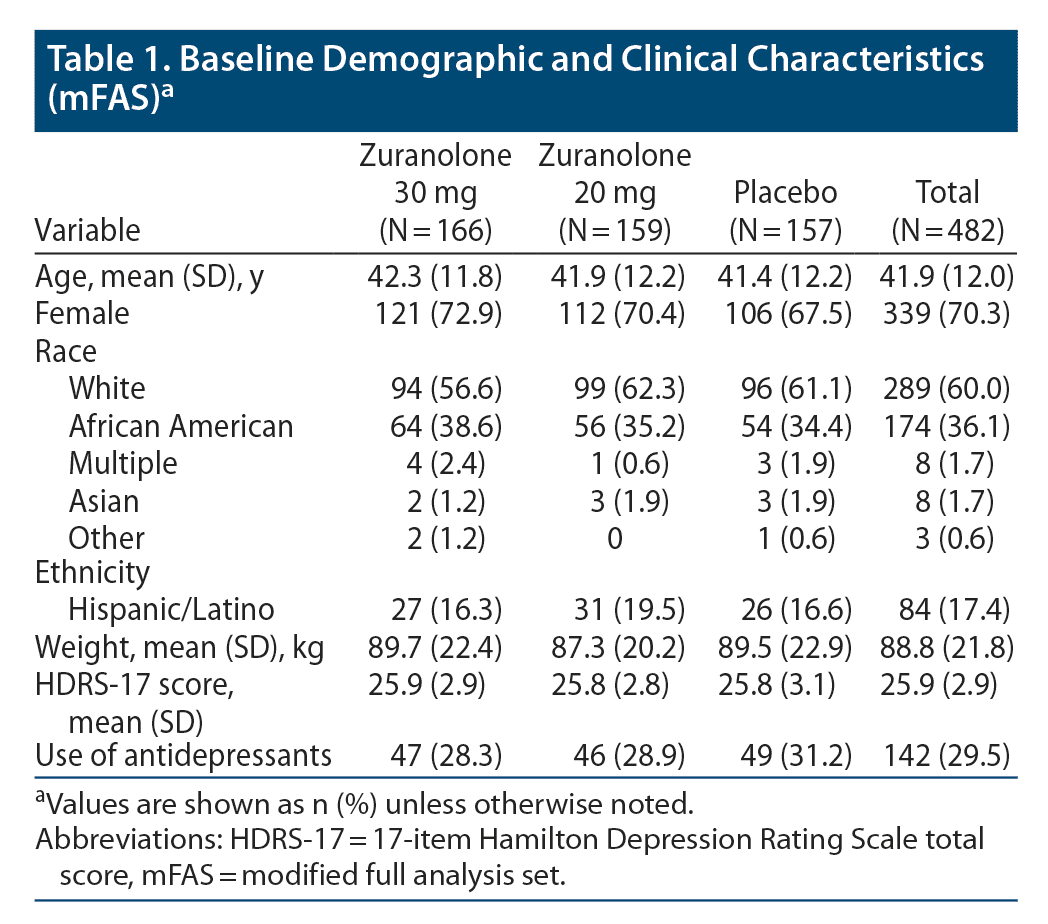
In the mFAS population, baseline demographic and clinical characteristics were well balanced among the treatment groups: most patients were female (70.3%) and White (60.0%); mean age was 41.9 years, and 29.5% were using antidepressants at baseline (Table 1). The median (range) time on treatment was 14 (1–18) days. Overall, 93.2% of patients received ≥ 11 doses of study drug, including 71.4% who received all 14 planned doses of study drug.
Primary Endpoint (mFAS)
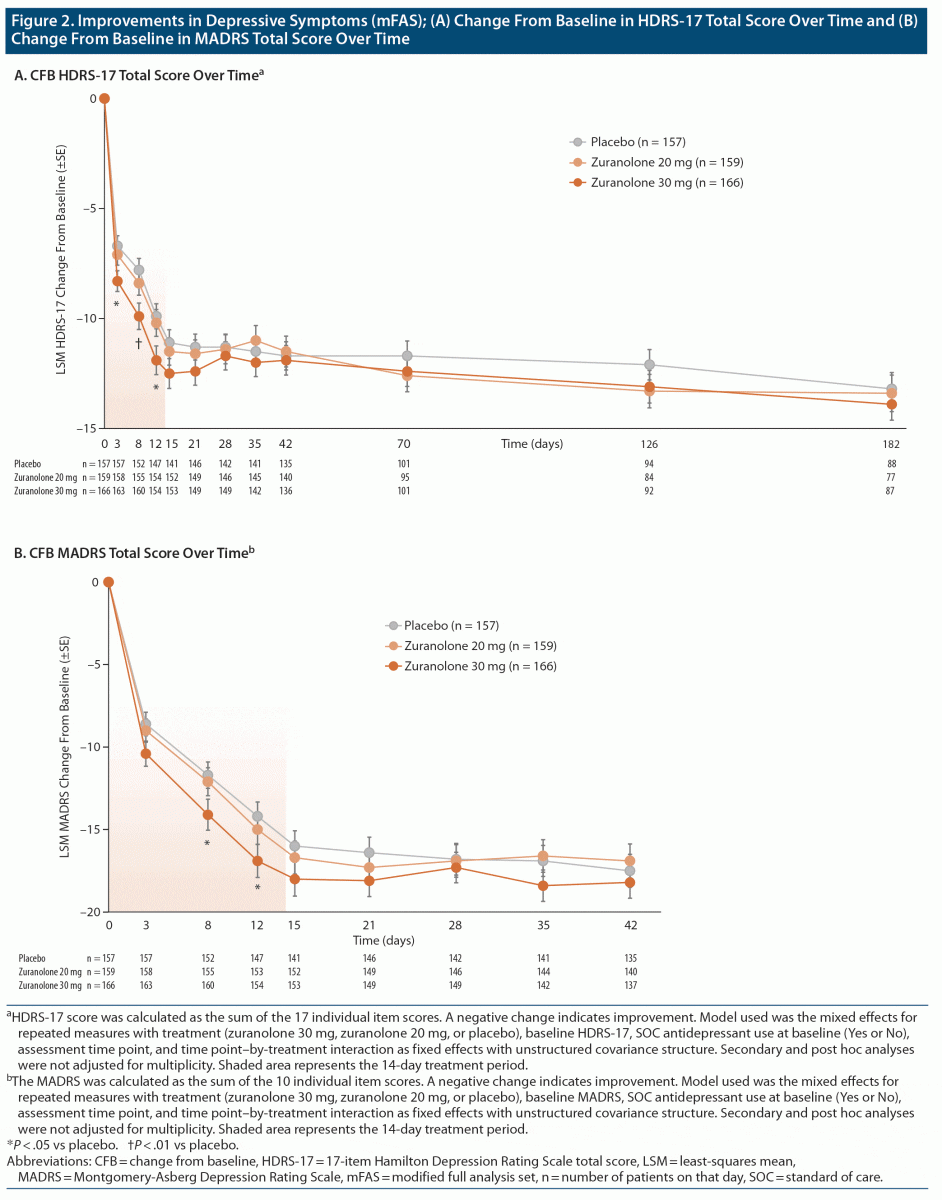
This study did not meet its primary endpoint; neither zuranolone 20 nor 30 mg vs placebo was associated with a significant CFB in HDRS-17 at day 15 (Figure 2A). Zuranolone 20 mg was associated with an LSM (SE) CFB at day 15 of −11.5 (0.62, LSM [SE] difference: −0.4 [0.85]; P = .664). Zuranolone 30 mg was associated with an LSM (SE) CFB at day 15 of −12.5 (0.68) vs −11.1 (0.59) for placebo (LSM [SE] difference: −1.4 [0.89]; P = .116). The Cohen d at day 15 was 0.03 for zuranolone 20 mg and 0.17 for zuranolone 30 mg (Supplementary Table 1).
Post Hoc Analyses
Among patients with more severe disease (HDRS-17 ≥ 24), post hoc analyses demonstrated a significant CFB in HDRS-17 at day 15 for patients receiving zuranolone 30 mg (n = 124; LSM [SE] −13.6 [0.8]) vs placebo (n = 115; −11.4 [0.71]; LSM [SE] difference: −2.3 [1.05], P = .032) (Figure 3A). The Cohen d at day 15 was 0.33 for patients with more severe disease receiving zuranolone 30 mg (Supplementary Table 2). Similarly, when patients with no measurable plasma zuranolone concentration (30/338; 8.9%) were excluded from the mFAS, a significant difference in the CFB in HDRS-17 at day 15 was observed for patients who received zuranolone 30 mg (n = 151; LSM [SE] −13.0 [0.72]) vs placebo (n = 157; −11.1 [0.59]; LSM [SE] difference: −1.8 [0.92], P = .049) (Figure 3B). The Cohen d at day 15 was 0.23 for patients with measurable plasma zuranolone concentration receiving zuranolone 30 mg (Supplementary Table 2). Among patients with both a baseline HDRS-17 ≥ 24 and measurable zuranolone concentration, LSM CFB at day 15 was significantly greater with zuranolone 30 mg (n = 115; LSM [SE] −13.9 [0.84]) vs placebo (n = 115; −11.4 [0.71]; LSM [SE] difference: −2.6 [1.08], P = .018) (Figure 3C). The Cohen d at day 15 was 0.32 for patients with a baseline HDRS-17 ≥ 24 and measurable plasma zuranolone concentration receiving zuranolone 30 mg (Supplementary Table 2). Patients who received zuranolone 20 mg did not show any significant differences from placebo at any assessment time point in these exploratory post hoc analyses.
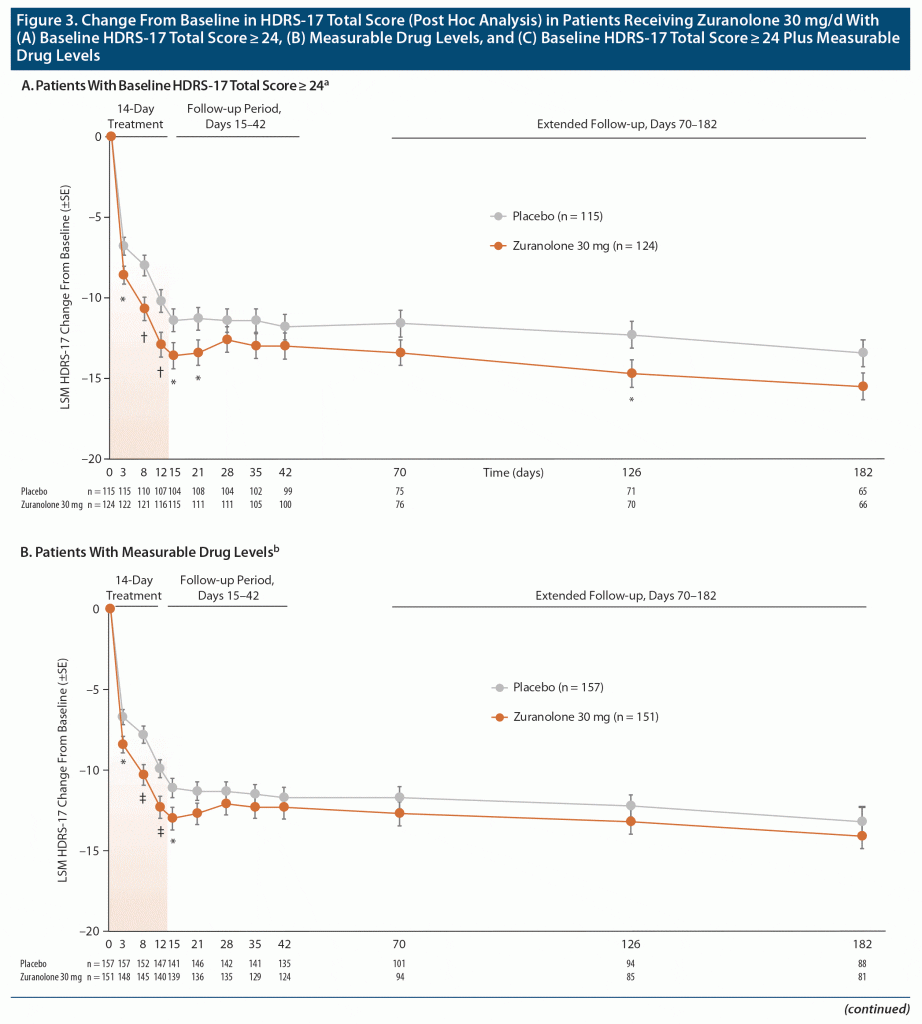
Secondary Endpoints (mFAS)
The LSM (SE) CFB in CGI-S at day 15 in the zuranolone 30 mg, zuranolone 20 mg, and placebo groups was −1.7 (0.11), −1.6 (0.11), and −1.5 (0.10), respectively. The LSM (SE) differences between zuranolone 30 and 20 mg vs placebo groups were −0.2 ([0.15]; P = .108) and −0.1 ([0.15]; P = .691), respectively.
A statistically significant between-group difference in LSM (SE) CFB in the HDRS-17 was noted for zuranolone 30 mg vs placebo at day 3 (−8.3 [0.47] vs −6.7 [0.46]; P = .016), day 8 (−9.9 [0.60] vs −7.8 [0.53]; P = .008), and day 12 (−11.9 [0.65] vs −9.9 [0.57]; P = .018) (Figure 2A). The Cohen d values for zuranolone 30 mg at days 3, 8, and 12 were 0.28, 0.26, and 0.26, respectively (Supplementary Table 1). No significant differences were seen for zuranolone 30 mg vs placebo at any time points after day 15. In the exploratory post hoc analyses, patients with more severe disease (HDRS-17 ≥ 24) showed a significant CFB in HDRS-17 at day 3 (P = .015), day 8 (P = .005), day 12 (P = .007), day 15 (P = .032), and day 21 (P = .048) (Figure 3A); patients with measurable plasma zuranolone concentration showed a significant CFB in HDRS-17 at day 3 (P = .012), day 8 (P = .003), day 12 (P = .007), and day 15 (P = .049) (Figure 3B); patients with both a baseline HDRS-17 ≥ 24 and measurable zuranolone concentration showed a significant CFB in HDRS-17 at day 3 (P = .016), day 8 (P = .003), day 12 (P = .005), day 15 (P = .018), and day 21 (P = .031) (Figure 3C).
HDRS-17 response rates with zuranolone 30 mg were significantly higher with zuranolone 30 mg vs placebo at day 8 (33.8% vs 23.0%; P = .024) and day 12 (43.5% vs 32.7%; P = .034) (Supplementary Figure 2). At day 15, HDRS-17 response rates were 50.3%, 42.8%, and 42.6% for zuranolone 30 mg, zuranolone 20 mg, and placebo, respectively (all P vs placebo > .05); at day 182, the response rates were 58.6% (51/87), 50.6% (39/77), and 58.0% (51/88), respectively (all P vs placebo > .05).
At day 15, HDRS-17 remission rates were 31.4%, 23.0%, and 23.4% for zuranolone 30 mg, zuranolone 20 mg, and placebo, respectively; the only statistically significant between-group difference was for day 12 zuranolone 30 mg vs placebo (Supplementary Figure 3); at day 182, the remission rates were 39.1% (34/87), 37.7% (29/77), and 36.4% (32/88), respectively.
The LSM (SE) CFB in MADRS at day 15 was −18.0 (1.0) for zuranolone 30 mg (P = .144), −16.7 (1.0) for zuranolone 20 mg (P = .599), and −16.0 (0.9) for placebo (Figure 2B). Zuranolone 30 mg was statistically significant from placebo at days 8 (P = .048) and 12 (P = .038). CGI-I response rates at day 15 were 55.3% for zuranolone 30 mg (P = .120 vs placebo), 47.0% for zuranolone 20 mg (P = .830), and 46.1% for placebo (Supplementary Figure 4). At day 12, CGI-I response rates for zuranolone 30 mg were significantly higher vs placebo (49% vs 37%; P = .026). There were no other statistically significant between-group differences in CGI-I responses at any other assessment time point. At day 15, the LSM CFB (SE) in HARS was −9.4 (0.5) for zuranolone 30 mg (P = .287 vs placebo), −9.1 (0.5) for zuranolone 20 mg (P = .502), and −8.7 (0.5) for placebo (Supplementary Figure 5).
Exploratory Efficacy Endpoints (mFAS)
Among female patients, the mean baseline CSFQ-14 was 31.7 and 33.7 for zuranolone 30 mg (n = 121) and placebo (n = 105), respectively, indicating, on average, the presence of sexual dysfunction (ie, CSFQ-14 scores ≤ 47); among male patients, corresponding scores were 42.1 (n = 45) and 40.9 (n = 51), respectively, indicating, on average, the absence (or near absence) of sexual dysfunction (ie, CSFQ-14 scores ≤ 41). No worsening of sexual dysfunction was observed during the study, and there were no significant differences between zuranolone 30 mg and placebo in LSM CFB CSFQ-14 in females or males at days 15, 28, and 42 (Supplementary Figure 6).
Safety/Tolerability
The percentage of patients with treatment-emergent adverse events (TEAEs) during the double-blind period (14-day treatment period and 4-week observation) was similar in the zuranolone 30 mg (54.7%), 20 mg (50.5%), and placebo (48.9%) groups (Table 2). The most common TEAEs (≥ 5% of patients in any group) for zuranolone 30 mg, zuranolone 20 mg, and placebo, respectively, were fatigue (6.8%, 1.6%, 2.6%), somnolence (6.8%, 5.9%, 4.2%), headache (6.3%, 11.2%, 7.4%), dizziness (5.7%, 7.4%, 3.7%), diarrhea (6.3%, 5.9%, 5.3%), sedation (4.7%, 5.9%, 3.2%), and nausea (3.6%, 5.3%, 4.7%). The incidence of weight increase or weight gain was low, with related adverse events being reported in 1 patient in the zuranolone 30-mg group, 1 in the zuranolone 20-mg group, and 4 in the placebo group.
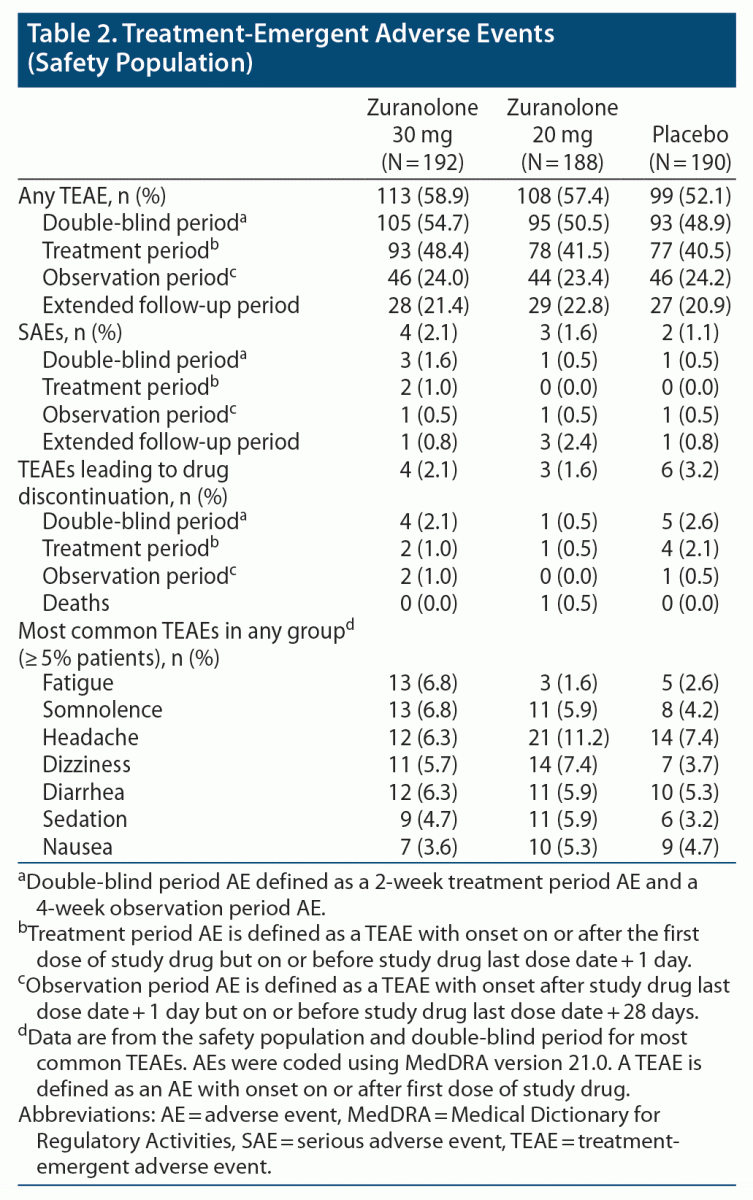
During the treatment period, 2 patients receiving zuranolone 30 mg experienced serious adverse events (SAEs): 1 suicide attempt on day 5 in a patient with a long-standing history of MDD and a past suicide attempt (possibly drug-related) and 1 bile duct stone on day 2 (requiring removal) in a patient with prior bile duct repair (not drug-related). During the follow-up observation period, SAEs were reported in 3 patients: 1 patient in the zuranolone 30-mg group, with syncope, ankle fracture, cervical vertebral fracture, and tibia fracture (day 28; not related); 1 patient in the zuranolone 20-mg group, with toxic encephalopathy, agitation, delirium, drug abuse, pneumonia, rhabdomyolysis, acute kidney injury, and respiratory failure (day 39; not related, deemed related to cocaine use); and 1 patient in the placebo group, with suicidal ideation (day 22; not related). TEAEs leading to treatment discontinuation were comparable across groups (zuranolone 30 mg: 2.1%, zuranolone 20 mg: 1.6%, and placebo: 3.2%). The most common reasons for withdrawal were psychiatric and nervous system disorders. No TEAEs of loss of consciousness were reported. No clinically significant changes in vital signs, clinical laboratory parameters, or electrocardiograms were observed. One patient in the zuranolone 20-mg group died during the 6-month extended follow-up period after treatment discontinuation (day 142; assessed as not treatment-related by the investigator) (see Supplementary Appendix 4 for details).
The percentages of patients experiencing suicidal ideation/behavior (assessed using C-SSRS) considerably decreased from baseline in all treatment groups. Fewer than 13% of patients in any group reported suicidal ideation/behavior from day 3 through the last assessment while on treatment. At day 182, there was no change in the percentage of patients reporting suicidal ideation/behavior in the zuranolone 30-mg (2.3% [3/131]), zuranolone 20-mg (4.7% [6/127]), or placebo (7.0% [9/129]) groups. The mean (SD) CFB PWC-20 total scores for zuranolone 30 mg, zuranolone 20 mg, and placebo were −5.7 (7.7), −5.4 (7.5), and −5.4 (7.0), respectively, at day 15, and −6.0 (7.0), −6.1 (7.3), and −5.6 (7.3), respectively, at day 21 (negative change indicates improvement).
DISCUSSION
The primary endpoint of the phase 3 MOUNTAIN study, CFB in HDRS-17 at day 15, was not met in either the 20-mg or 30-mg treatment group. However, results from this study may indicate that zuranolone represents a novel approach to treating patients with MDD. Patients receiving zuranolone experienced a rapid onset of improvement in depressive symptoms as early as day 3 after beginning a 14-day treatment course. Although not significantly different from the placebo group, the HDRS-17 response rates in patients receiving zuranolone 30 mg were similar between day 15 (50.3%) and day 182 (58.6%).
Nonadherence with antidepressant therapy is a considerable problem in patients with MDD. Some patients discontinue treatment because of lack of effect, residual symptoms, or adverse effects or from depression itself.61–63 Common residual symptoms from antidepressants include blunted affect, sleep disturbances, weight gain, sexual dysfunction, cognitive impairments, and fatigue.28,64–67 Zuranolone was generally well tolerated, with a safety and tolerability profile in patients with MDD consistent with an earlier phase 2 study.54
In the phase 2 study, significant improvements in depressive symptoms were observed through day 28 with zuranolone 30 mg vs placebo. Although greater numerical improvements in depressive symptoms were observed with zuranolone 30 mg vs placebo at all measured time points in the current study, they only reached significance beyond day 12 in patients with detectable plasma zuranolone concentration and/or more severe disease (HDRS-17 total score ≥ 24) at baseline. Undetectable plasma zuranolone concentrations could indicate nonadherence. In this study, patients were treated as outpatients during their clinic visits on days 1, 3, 8, and 12. Patients were required to take a video of drug ingestion at home. Study drug adherence was defined as the number of doses taken, divided by the number of doses planned to be taken (14), times 100; 93.2% of patients received ≥ 11 of the planned 14 doses, and 71.4% of patients received all 14 doses of study drug. Overall, adherence to study drug was 98.3%, and there was no notable difference among the treatment groups. In contrast, patients in the phase 2 trial received treatment as inpatients on days 1–7 and as inpatients or during outpatient residential or clinic visits on days 8–14, increasing the likelihood that patients received all planned doses of study drug.
It is recognized that the large placebo effects observed in many studies of antidepressant therapies may contribute to nonsignificant treatment differences in those studies. There was also a robust placebo response in this phase 3 trial. This may be attributable to the high number of patient visits in this study (10 visits in 42 days) compared to the number of visits in the real-world setting. It is hypothesized that frequent supervision and assessment of patients in clinical trials of depression contribute to the placebo effect.68,69 An exploratory analysis of efficacy data from approximately 80 placebo-controlled MDD trials reported diminishing treatment effect size between 1983 and 2008 in both US and non-US trials.70 The magnitude of the effect size depends on the placebo response and may explain the relatively low effect size observed for the primary endpoint in the MOUNTAIN Study. However, for the post hoc analyses, the Cohen d estimates were greater than 0.2, a threshold considered clinically meaningful.71,72 Nevertheless, the trend toward improvement observed here warrants additional research in studies exploring potentially higher doses of zuranolone.
This study had some limitations. As with most placebo-controlled clinical trials that use stringent inclusion and exclusion criteria in an attempt to enroll a homogeneous patient population, the results reported here may not be generalizable to all patients with MDD. In addition, the need to amend the eligibility criteria in the middle of the study to ensure that the study enrolled patients with the intended severity of depression and the fact that approximately 9% of patients taking zuranolone 30 mg did not have detectable plasma zuranolone concentration could potentially limit the interpretation of the study outcomes reported here. While this study included an extended follow-up period of 6 months, patients did not have the option to receive repeat treatment courses of zuranolone if a subsequent depressive episode occurred, limiting our ability to use these data to inform real-world, long-term clinical use. The results from other completed and ongoing studies of zuranolone in patients with MDD have demonstrated a significant improvement in depressive symptoms with a 14-day treatment course.54,73 While patients taking antidepressants at baseline were required to remain on the stable dose through day 42, there was no requirement to remain on the stable dose of antidepressants after day 42 (during the 6-month extended follow-up period). Subsequent studies within the LANDSCAPE and NEST clinical development programs have been designed to address some of these limitations: using a higher dose of zuranolone (50 mg), enrolling more diverse patient populations (eg, extending the upper age limit to 75 years), and including the option of repeat treatment courses as needed.
CONCLUSIONS
The phase 3 MOUNTAIN study did not meet its primary endpoint: results did not show a significant improvement vs placebo in depressive symptoms in adult patients with MDD receiving zuranolone, as assessed by CFB in HDRS-17 total score at day 15.
However, significant improvements in depressive symptoms vs placebo were observed with zuranolone 30 mg as early as day 3, and at days 8 and 12. In post hoc analyses of patients with more severe disease and/or measurable plasma concentration, zuranolone 30 mg again separated from placebo at day 3, and significant improvements were observed through day 15. Zuranolone was generally well tolerated and demonstrated a safety profile similar to that observed in earlier studies. Zuranolone continues to be evaluated as an oral, rapid-onset, 14-day treatment course for patients with MDD.
Submitted: March 7, 2022; accepted November 10, 2022.
Published online: February 20, 2023.
Relevant financial relationships: Dr Clayton reports receiving research grants from Daré Bioscience, Janssen, Otsuka, Praxis Precision Medicines, Relmada Therapeutics, Inc., and Sage Therapeutics, Inc.; consulting fees from AbbVie, Inc., Brii Biosciences, Fabre-Kramer, Janssen Research & Development, LLC, Mind Cure Health, Ovoca Bio plc, Praxis Precision Medicine, PureTech Health, Reunion Neuroscience (formerly Field Trip Health), S1 Biopharma, Sage Therapeutics, Inc., Takeda/Lundbeck, Vella Bioscience, Inc., and WCG MedAvante-ProPhase; royalties from Ballantine Books/Random House, the Changes in Sexual Functioning Questionnaire, and Guilford Publications; and restricted stock in Euthymics, Mediflix LLC, and S1 Biopharma. Drs Lasser, Nandy, Sankoh, and Jonas are employees of Sage Therapeutics, Inc., and hold stock or stock options. Dr Kanes is a former employee of Sage Therapeutics, Inc., and holds stock. He is presently an employee of Ancora Bio.
Funding/support: This trial was funded by Sage Therapeutics, Inc.
Role of the sponsor: The study sponsor was involved in the study design, data analysis, interpretation, and writing of the report. Biogen Inc. had the opportunity to review the manuscript. The authors had full editorial control of the manuscript and provided their final approval on all content.
Previous presentations: Aspects of this study have been presented at the Society of Biological Psychiatry, Virtual Annual Meeting, April 30–May 2, 2020; the American Academy of Neurology Annual Meeting, April 25–May 1, 2020, Toronto, Canada; the World Congress of Psychiatry, Virtual Annual Meeting, March 10–13, 2021; and the American Society of Clinical Psychopharmacology, Virtual Annual Meeting, June 1–4, 2021.
Acknowledgments: Medical writing and editorial assistance were provided by Sean Sheffler-Collins, PhD, and Leonard Lionnet, PhD, of Symbiotix, LLC, and were funded by Sage Therapeutics, Inc., and Biogen Inc. The authors received no honorarium/fee or other form of financial support related to the development of this article. Brian Werneburg, PhD, and Handan Gunduz-Bruce, MD, MBA, are former employees of Sage Therapeutics, Inc., and hold stock or stock options. The authors thank Brian Werneburg and Handan Gunduz-Bruce for their contributions to this study, which included critical input for study conception and design, and data analysis and interpretation.
Supplementary material: Available at Psychiatrist.com.
CLINICAL POINTS
- Standard-of-care antidepressants are often associated with treatment-limiting adverse effects and can require weeks or months to produce effects, potentially resulting in negative outcomes, including decreased likelihood of remission and nonadherence.
- A short-term course of a monotherapy or adjunctive treatment that leads to rapid response, is generally well tolerated, and maintains effect over 6 months would be a paradigm shift in treatment options.
Author Affiliations

- University of Virginia School of Medicine, Charlottesville, Virginia
- Corresponding author: Anita H. Clayton, MD, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia School of Medicine, PO Box 800623, Charlottesville, VA 22908-0623 ([email protected]).
- Sage Therapeutics, Inc, Cambridge, Massachusetts
- Sage Therapeutics, Inc, Cambridge, Massachusetts
- Sage Therapeutics, Inc, Cambridge, Massachusetts
- Sage Therapeutics, Inc, Cambridge, Massachusetts
- Sage Therapeutics, Inc, Cambridge, Massachusetts
References (73)
- 2019 NSDUH Detailed Tables. 2019 National Survey of Drug Use and Health (NSDUH). SAMHSA website. https://www.samhsa.gov/data/report/2019-nsduh-detailed-tables. September 11, 2020. Cited December 15, 2020.
- Kessler RC, Berglund P, Demler O, et al; National Comorbidity Survey Replication. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–3105. PubMed CrossRef
- Mokdad AH, Ballestros K, Echko M, et al; US Burden of Disease Collaborators. The state of US health, 1990-2016: burden of diseases, injuries, and risk factors among US states. JAMA. 2018;319(14):1444–1472. PubMed CrossRef
- World Health Organization. Global Health Estimates 2019: disease burden by cause, age, sex, by country, and by region, 2000–2019. WHO website. https://www.who.int/docs/default-source/gho-documents/global-health-estimates/ghe2019_yld_global_2000_2019c417f68b-841d-4a7a-9e5c-f087f9f86e48.xlsx?sfvrsn=dac29788_7. Cited December 2020.
- Greenberg PE, Fournier AA, Sisitsky T, et al. The economic burden of adults with major depressive disorder in the United States (2005 and 2010). J Clin Psychiatry. 2015;76(2):155–162. PubMed CrossRef
- Gelenberg AJ, Freeman MP, Markowitz JC. American Psychiatric Association Practice Guideline for the treatment of patients with major depressive disorder. 3rd ed. Am J Psychiatry. 2010;167:1–152.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5). 5th ed. American Psychiatric Association; 2013.
- Batterham PJ, Christensen H, Mackinnon AJ. Modifiable risk factors predicting major depressive disorder at four year follow-up: a decision tree approach. BMC Psychiatry. 2009;9(1):75. PubMed CrossRef
- Fischer AS, Keller CJ, Etkin A. The clinical applicability of functional connectivity in depression: pathways toward more targeted intervention. Biol Psychiatry Cogn Neurosci Neuroimaging. 2016;1(3):262–270. PubMed CrossRef
- Malhi GS, Mann JJ. Depression. Lancet. 2018;392(10161):2299–2312. PubMed CrossRef
- Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130(2):226–238. PubMed CrossRef
- Schiller CE, Johnson SL, Abate AC, et al. Reproductive steroid regulation of mood and behavior. Compr Physiol. 2016;6(3):1135–1160. PubMed CrossRef
- Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16(4):383–406. PubMed CrossRef
- Sarawagi A, Soni ND, Patel AB. Glutamate and GABA homeostasis and neurometabolism in major depressive disorder. Front Psychiatry. 2021;12:637863. PubMed CrossRef
- Fogaça MV, Duman RS. Cortical GABAergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci. 2019;13:87. PubMed CrossRef
- Gabbay V, Mao X, Klein RG, et al. Anterior cingulate cortex γ-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch Gen Psychiatry. 2012;69(2):139–149. PubMed CrossRef
- Gold BI, Bowers MB Jr, Roth RH, et al. GABA levels in CSF of patients with psychiatric disorders. Am J Psychiatry. 1980;137(3):362–364. PubMed CrossRef
- Hasler G, van der Veen JW, Tumonis T, et al. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2007;64(2):193–200. PubMed CrossRef
- Lener MS, Niciu MJ, Ballard ED, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. 2017;81(10):886–897. PubMed CrossRef
- Maguire J. Neuroactive steroids and GABAergic involvement in the neuroendocrine dysfunction associated with major depressive disorder and postpartum depression. Front Cell Neurosci. 2019;13:83. PubMed CrossRef
- Petty F, Kramer GL, Gullion CM, et al. Low plasma gamma-aminobutyric acid levels in male patients with depression. Biol Psychiatry. 1992;32(4):354–363. PubMed CrossRef
- Sanacora G, Mason GF, Rothman DL, et al. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1999;56(11):1043–1047. PubMed CrossRef
- Guilloux JP, Douillard-Guilloux G, Kota R, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17(11):1130–1142. PubMed CrossRef
- Rajkowska G, O’Dwyer G, Teleki Z, et al. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32(2):471–482. PubMed CrossRef
- Sibille E, Wang Y, Joeyen-Waldorf J, et al. A molecular signature of depression in the amygdala. Am J Psychiatry. 2009;166(9):1011–1024. PubMed CrossRef
- Fee C, Banasr M, Sibille E. Somatostatin-positive gamma-aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry. 2017;82(8):549–559. PubMed CrossRef
- Levinson AJ, Fitzgerald PB, Favalli G, et al. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. 2010;67(5):458–464. PubMed CrossRef
- Northoff G, Sibille E. Why are cortical GABA neurons relevant to internal focus in depression? a cross-level model linking cellular, biochemical and neural network findings. Mol Psychiatry. 2014;19(9):966–977. PubMed CrossRef
- Duman RS, Sanacora G, Krystal JH. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102(1):75–90. PubMed CrossRef
- Lam RW, McIntosh D, Wang J, et al; CANMAT Depression Work Group. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder, Section 1: disease burden and principles of care. Can J Psychiatry. 2016;61(9):510–523. PubMed CrossRef
- Stahl SM. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 4th ed. Cambridge University Press; 2013.
- Hockenberry JM, Joski P, Yarbrough C, et al. Trends in treatment and spending for patients receiving outpatient treatment of depression in the United States, 1998–2015. JAMA Psychiatry. 2019;76(8):810–817. PubMed CrossRef
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. PubMed CrossRef
- Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. PubMed CrossRef
- Henkel V, Seemüller F, Obermeier M, et al. Does early improvement triggered by antidepressants predict response/remission? analysis of data from a naturalistic study on a large sample of inpatients with major depression. J Affect Disord. 2009;115(3):439–449. PubMed CrossRef
- Hennings JM, Owashi T, Binder EB, et al. Clinical characteristics and treatment outcome in a representative sample of depressed inpatients - findings from the Munich Antidepressant Response Signature (MARS) project. J Psychiatr Res. 2009;43(3):215–229. PubMed CrossRef
- Jakubovski E, Bloch MH. Prognostic subgroups for citalopram response in the STAR*D trial. J Clin Psychiatry. 2014;75(7):738–747. PubMed CrossRef
- Ashton AK, Jamerson BD, L Weinstein W, et al. Antidepressant-related adverse effects impacting treatment compliance: results of a patient survey. Curr Ther Res Clin Exp. 2005;66(2):96–106. PubMed CrossRef
- Demyttenaere K, Enzlin P, Dewé W, et al. Compliance with antidepressants in a primary care setting, 1: beyond lack of efficacy and adverse events. J Clin Psychiatry. 2001;62(suppl 22):30–33. PubMed
- Fortney JC, Pyne JM, Edlund MJ, et al. Reasons for antidepressant nonadherence among veterans treated in primary care clinics. J Clin Psychiatry. 2011;72(6):827–834. PubMed CrossRef
- Oluboka OJ, Katzman MA, Habert J, et al. Functional recovery in major depressive disorder: providing early optimal treatment for the individual patient. Int J Neuropsychopharmacol. 2018;21(2):128–144. PubMed CrossRef
- A study to evaluate SAGE-217 in adult participants with major depressive disorder (MDD). ClinicalTrials.gov identifier: NCT03864614. ClinicalTrials.gov website. https://ClinicalTrials.gov/show/NCT03864614. February 9, 2021. Cited March 4, 2021.
- A comparative study of Sage-217 plus an antidepressant (ADT) versus placebo plus an ADT in adults with major depressive disorder. ClinicalTrials.gov identifier: NCT04476030. Clinical Trials.gov website. https://ClinicalTrials.gov/show/NCT04476030. February 23, 2021 Cited March 4, 2021.
- Hoffmann E, Nomikos GG, Kaul I, et al. SAGE-217, A novel GABA(A) receptor positive allosteric modulator: clinical pharmacology and tolerability in randomized phase I dose-finding studies. Clin Pharmacokinet. 2020;59(1):111–120. PubMed CrossRef
- Martinez Botella G, Salituro FG, Harrison BL, et al. Neuroactive steroids. 2. 3alpha-hydroxy-3beta-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5beta-pregnan-20 -one (SAGE-217): a clinical next generation neuroactive steroid positive allosteric modulator of the (gamma-aminobutyric acid)A receptor. J Med Chem. 2017;60(18):7810–7819. PubMed CrossRef
- Althaus AL, Ackley MA, Belfort GM, et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology. 2020;181:108333. PubMed CrossRef
- Reddy DS, Estes WA. Clinical potential of neurosteroids for CNS Disorders. Trends Pharmacol Sci. 2016;37(7):543–561. PubMed CrossRef
- Sugasawa Y, Bracamontes JR, Krishnan K, et al. The molecular determinants of neurosteroid binding in the GABA(A) receptor. J Steroid Biochem Mol Biol. 2019;192:105383. PubMed CrossRef
- Sugasawa Y, Cheng WW, Bracamontes JR, et al. Site-specific effects of neurosteroids on GABAA receptor activation and desensitization. eLife. 2020;9:eLife.55331. PubMed
- Ziolkowski L, Mordukhovich I, Chen DM, et al. A neuroactive steroid with a therapeutically interesting constellation of actions at GABAA and NMDA receptors. Neuropharmacology. 2021;183:108358. PubMed CrossRef
- Abramian AM, Comenencia-Ortiz E, Modgil A, et al. Neurosteroids promote phosphorylation and membrane insertion of extrasynaptic GABAA receptors. Proc Natl Acad Sci U S A. 2014;111(19):7132–7137. PubMed CrossRef
- Modgil A, Parakala ML, Ackley MA, et al. Endogenous and synthetic neuroactive steroids evoke sustained increases in the efficacy of GABAergic inhibition via a protein kinase C-dependent mechanism. Neuropharmacology. 2017;113(Pt A):314–322. PubMed CrossRef
- Parakala ML, Zhang Y, Modgil A, et al. Metabotropic, but not allosteric, effects of neurosteroids on GABAergic inhibition depend on the phosphorylation of GABAA receptors. J Biol Chem. 2019;294(32):12220–12230. PubMed CrossRef
- Gunduz-Bruce H, Silber C, Kaul I, et al. Trial of SAGE-217 in patients with major depressive disorder. N Engl J Med. 2019;381(10):903–911. PubMed CrossRef
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62. PubMed CrossRef
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. PubMed CrossRef
- Guy W. ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976.
- Keller A, McGarvey EL, Clayton AH. Reliability and construct validity of the Changes in Sexual Functioning Questionnaire short-form (CSFQ-14). J Sex Marital Ther. 2006;32(1):43–52. PubMed CrossRef
- Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. PubMed CrossRef
- Rickels K, Garcia-Espana F, Mandos LA, et al. Physician Withdrawal Checklist (PWC-20). J Clin Psychopharmacol. 2008;28(4):447–451. PubMed CrossRef
- Grenard JL, Munjas BA, Adams JL, et al. Depression and medication adherence in the treatment of chronic diseases in the United States: a meta-analysis. J Gen Intern Med. 2011;26(10):1175–1182. PubMed CrossRef
- Rossom RC, Shortreed S, Coleman KJ, et al. Antidepressant adherence across diverse populations and healthcare settings. Depress Anxiety. 2016;33(8):765–774. PubMed CrossRef
- Samples H, Mojtabai R. Antidepressant self-discontinuation: results from the collaborative psychiatric epidemiology surveys. Psychiatr Serv. 2015;66(5):455–462. PubMed CrossRef
- Nierenberg AA, Husain MM, Trivedi MH, et al. Residual symptoms after remission of major depressive disorder with citalopram and risk of relapse: a STAR*D report. Psychol Med. 2010;40(1):41–50. PubMed CrossRef
- Romera I, Pérez V, Ciudad A, et al. Residual symptoms and functioning in depression, does the type of residual symptom matter? a post-hoc analysis. BMC Psychiatry. 2013;13(1):51. PubMed CrossRef
- Santarsieri D, Schwartz TL. Antidepressant efficacy and side-effect burden: a quick guide for clinicians. Drugs Context. 2015;4:212290. PubMed CrossRef
- Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a meta-analysis. J Clin Psychopharmacol. 2009;29(3):259–266. PubMed CrossRef
- Walsh BT, Seidman SN, Sysko R, et al. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840–1847. PubMed CrossRef
- Khan A, Fahl Mar K, Faucett J, et al. Has the rising placebo response impacted antidepressant clinical trial outcome? data from the US Food and Drug Administration 1987-2013. World Psychiatry. 2017;16(2):181–192. PubMed CrossRef
- Khin NA, Chen YF, Yang Y, et al. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. 2011;72(4):464–472. PubMed CrossRef
- McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo-controlled study of vortioxetine on cognitive function in depressed adults. Int J Neuropsychopharmacol. 2014;17(10):1557–1567. PubMed CrossRef
- Lund K, Vase L, Petersen GL, et al. Randomised controlled trials may underestimate drug effects: balanced placebo trial design. PLoS One. 2014;9(1):e84104. PubMed CrossRef
- Clayton A. Zuranolone in Major Depressive Disorder: Results From the Phase 3, Multicenter, Randomized, Double-Blind, Placebo-Controlled WATERFALL Study. Lisbon, Portugal: European College of Neuropsychopharmacology; 2021.
![]() Save
Save
![]() Share
Share
![]() Cite
Cite